Les encéphalopathies spongiformes (maladies à prions) sont les maladies dans lesquelles des formes pathologiques de protéines prions sont impliquées dans le développement. Nous en savons de plus en plus sur les maladies à prions, mais les aspects clés restent inconnus - à l'heure actuelle, la médecine n'a pas les moyens de guérir les patients de ces maladies.
Les encéphalopathies spongiformes, ou maladies à prions, peuvent se développer au cours de la vie, tandis que d'autres résultent de mutations génétiques héréditaires présentes dès la naissance. Au sein de ce groupe, il existe plusieurs entités présentes chez l'homme, par exemple la maladie de Creutzfeldt-Jakob ou l'insomnie familiale mortelle.
Les maladies à prions sont depuis longtemps très mystérieuses. Contrairement à d'autres agents pathogènes, tels que les bactéries, les virus ou les champignons, ils ne contiennent pas d'acide nucléique - les prions sont uniquement constitués de protéines. La théorie des maladies à prions a été découverte par S. Prusiner, cette découverte a été très appréciée dans la communauté scientifique - en 1997, le chercheur a reçu le prix Nobel de médecine. Bien que relativement de nombreuses années se soient écoulées depuis la naissance du concept de prion, certains scientifiques pensent toujours qu'il est incomplet et étudient plus en détail la nature de ces conditions - certains des facteurs responsables des encéphalopathies spongiformes ont maintenant été confirmés.
Maladies à prions: causes
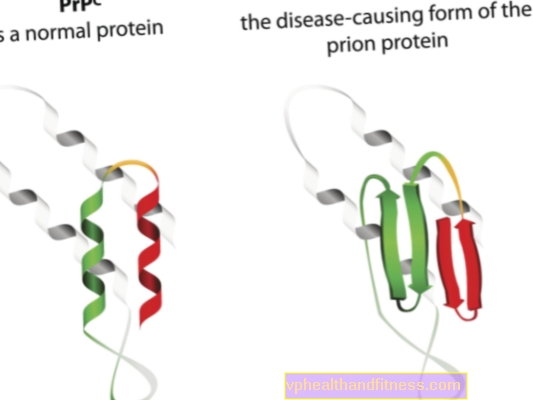
L'étiologie des maladies à prions est liée à la transformation des protéines prions normales en formes pathogènes et pathogènes. Les prions sont des molécules de protéines présentes dans le corps de chaque être humain. Leur fonction n'est pas encore tout à fait claire, mais on sait que dans des conditions normales, les protéines prions ne nuisent pas à l'organisme. La situation est différente lorsque les prions changent de structure et deviennent des particules pathogènes - alors l'une des nombreuses encéphalopathies spongiformes se développe. Les prions naturellement présents dans le corps sont appelés PRPC, tandis que les formes anormales sont appelées PRPSC. Ces derniers sont un problème sérieux non seulement parce qu'ils peuvent s'accumuler dans le tissu nerveux sous forme de dépôts et lui causer des dommages, mais aussi parce qu'ils ont la capacité de transformer des prions normaux en une forme malformée (en termes simples, PRPSC peut «infecter» les protéines normales avec son potentiel pathogène).
Lisez aussi: Maladie de Huntington (chorée de Huntington): causes, symptômes, traitement Tremblements musculaires - causes. Que signifie le tremblement musculaire? Maladies qui tuent le plus vite: CHOC, EBOLA, DAMN, ATTAQUE, URGENCE [GALE ...Fondamentalement, il existe 3 causes d'encéphalopathies spongiformes:
- sporadique (une mutation pathogène se produit dans les cellules somatiques, elle survient au cours de la vie du patient),
- famille (résultant du fardeau des mutations héritées des parents),
- Passage (lié à l'introduction de prions pathogènes dans le corps humain, par exemple à travers des préparations d'hormones de croissance contaminées par ces particules ou une transplantation cornéenne d'une personne souffrant d'une encéphalopathie spongiforme).
Encéphalopathies spongiformes: maladie de Creutzfeldt-Jakob
La maladie de Creutzfeldt-Jakob (MCJ) a été décrite pour la première fois au début des années 1920. Il existe 4 types de maladies:
- MCJ sporadique (la plus courante, représentant jusqu'à 9/10 de tous les cas de MCJ)
- ville natale de la MCJ
- submergé par la MCJ
- variante de la MCJ
Le tableau clinique au cours de diverses variantes de la maladie de Creutzfeldt-Jakob peut être variable. Les affections les plus courantes au cours de ce groupe d'encéphalopathies spongiformes sont:
- troubles de la démence (y compris détérioration progressive de la mémoire, de l'attention et de la concentration)
- myoclonie (mouvements involontaires comme des secousses soudaines des muscles)
- dysfonctionnement cérébelleux (se manifestant, par exemple, par des troubles de l'équilibre)
- Vision floue
- symptômes pyramidaux et extrapyramidaux
Au cours des variantes de la MCJ, des troubles mentaux (par exemple, anxiété, humeur dépressive), douleur et autres mouvements involontaires autres que ceux mentionnés ci-dessus peuvent également apparaître.
Le pronostic de la maladie de Creutzfeldt-Jakob est médiocre - par exemple, chez les patients atteints de MCJ sporadique, il faut en moyenne quatre à cinq mois entre le début des symptômes de la maladie et le décès.
Encéphalopathies spongiformes: syndrome de Gerstmann-Straussler-Scheinker
Le syndrome de Gerstmann-Straussler-Scheinker (GSS) se déroule généralement dans les familles et est causé par une mutation héréditaire du gène PRNP. Elle est considérée comme l'encéphalopathie spongiforme à progression la plus lente. L'équipe GSS comprend:
- ataxie spinocérébelleuse
- dysarthrie
- troubles de la démence
- troubles de la déglutition
- nystagmus
- augmentation de la tension musculaire
Les patients diagnostiqués avec un GSS ont une durée variable et, chez certains patients, la mort survient plus de 10 ans après son apparition.
Encéphalopathies spongiformes: insomnie familiale mortelle
L'insomnie familiale fatale est une maladie à prions causée par des mutations du gène PRNP. La maladie est extrêmement rare et a jusqu'à présent été diagnostiquée dans 28 familles dans le monde. Au cours de l'insomnie familiale fatale, le premier symptôme est l'incapacité de dormir. Ce problème entraîne des troubles anxieux et le patient a des hallucinations. L'effet du manque constant de repos nocturne est un dysfonctionnement du système autonome (y compris des modifications de la fonction cardiaque, de la transpiration et des troubles du système digestif), il y a également une diminution progressive du poids corporel. Dans les stades plus avancés de l'insomnie familiale fatale, des troubles hormonaux apparaissent et des symptômes de démence surviennent au cours de la maladie.
Le pronostic de l'insomnie familiale fatale, comme pour les autres encéphalopathies spongiformes, est mauvais: les patients meurent généralement dans les trois ans suivant leur apparition.
Encéphalopathies spongiformes: prionopathie à sensibilité variable à la protéase
La survenue des encéphalopathies spongiformes discutées est principalement liée à des mutations dans le gène PRNP. Cependant, ces mutations concernent différents codons de ce gène, et par conséquent plusieurs maladies à prions différentes sont distinguées. Une unité décrite relativement récemment (en 2008) est la prionopathie avec une sensibilité variable à la protéase. Les personnes souffrant de cette maladie portent des mutations dans jusqu'à trois codons du gène PRNP.
Dans la prionopathie à sensibilité variable aux protéases, les patients présentent:
- déficience cognitive
- extrême gravité des troubles psychiatriques: ils peuvent être une euphorie et une agitation, mais aussi une apathie importante
- dysarthrie
- aphasie (troubles du langage)
La durée moyenne de la maladie dans cette prionopathie est inférieure à 4 ans.
Encéphalopathies spongiformes: kuru
Kuru est maintenant considérée comme une maladie qui n'existe pratiquement plus - elle a été trouvée chez des représentants de tribus de Papouasie-Nouvelle-Guinée, qui pratiquaient un comportement cannibale. Le symptôme dominant de cette encéphalopathie spongiforme est l'ataxie cérébelleuse progressive. Elle peut s'accompagner de mouvements involontaires (principalement sous forme de chorée, de tremblements et d'athétose), ainsi que d'incontinence urinaire et fécale. Les patients sous kuru éprouvent également des sautes d'humeur importantes, ils développent des réflexes primitifs (par exemple, la succion). Un problème tout à fait caractéristique dans le cas de cette maladie à prions sont des accès forcés de pleurs ou de rires - en raison de ce dernier phénomène, le kuru est parfois appelé «mort riante».
Encéphalopathies spongiformes: diagnostic
Les maladies à prions peuvent être suspectées sur la base des symptômes du patient. Cependant, ils sont assez non spécifiques, car ils peuvent également apparaître au cours d'un certain nombre d'autres maladies qui ne sont pas liées aux prions. Pour cette raison, les éléments suivants sont également utilisés dans le diagnostic des encéphalopathies spongiformes:
- des tests d'imagerie (ex: imagerie par résonance magnétique, qui permet de détecter les changements liés à la dégénérescence du cerveau par les protéines prions),
- des tests de laboratoire (tels que l'évaluation des concentrations de protéines dans le liquide céphalo-rachidien, par exemple les protéines MAP-tau, S-100 ou 14-3-3),
- tests génétiques (pour détecter la présence de mutations chez le patient),
- tests immunhistochimiques (utilisant des anticorps dirigés contre les protéines prion).
Le diagnostic peut également être confirmé par une autopsie du cerveau, dans laquelle il est possible de trouver des changements caractéristiques des encéphalopathies spongiformes. Il peut s'agir de lésions spongieuses, réparties de manière variée et de structure différente (en fonction de l'entité spécifique de la maladie), des plaques amyloïdes et des anomalies neuronales.
Encéphalopathies spongiformes: traitement
Les maladies à prions sont actuellement incurables - malgré de nombreuses études en cours depuis de nombreuses années, la médecine ne contient toujours pas de médicaments qui pourraient ralentir ou inhiber complètement leur progression. Le traitement symptomatique est utilisé chez les patients atteints d'encéphalopathies spongiformes, qui vise à atténuer l'intensité des symptômes et à améliorer autant que possible leur qualité de vie.
Cependant, les travaux sur le traitement des encéphalopathies spongiformes sont toujours en cours. Les scientifiques essaient d'utiliser diverses méthodes - le premier exemple est la thérapie génique. Ils affecteraient les acides nucléiques et les mutations présentes dans leur structure - le but de l'application de la thérapie génique serait de neutraliser les erreurs dans le code génétique. Une autre approche est à la base de l'immunothérapie - des travaux sont en cours pour créer des anticorps dont le rôle serait d'éliminer les prions pathogènes. Une autre méthode qui voit le potentiel pour lutter contre les encéphalopathies spongiformes est le traitement avec l'utilisation de molécules de protéines synthétisées, qui, une fois introduites dans le corps du patient, neutraliseraient les protéines pathologiques.
Article recommandé:
Encéphalopathies - causes, types et symptômes--przyczyny-objawy-i-leczenie.jpg)